

Paroxetine, aka Paxil aka Seroxat, is an SSRI antidepressant. Like other SSRIs, its reputation has see-sawed over time. Hailed as miracle drugs in the 1990s and promoted for everything from depression to "separation anxiety" in dogs, they fell from grace over the past decade.
Like other SSRIs, its reputation has see-sawed over time. Hailed as miracle drugs in the 1990s and promoted for everything from depression to "separation anxiety" in dogs, they fell from grace over the past decade.
First, concerns emerged over withdrawal symptoms and suicidality especially in young people. Then more recently their antidepressant efficacy came into serious question. Paroxetine has arguably the worst image of all SSRIs, although whether it's much different to the rest is unclear.
Now a new paper claims to provide a definitive assessment of the safety and efficacy of paroxetine in adults (age 18+). The lead authors are from GlaxoSmithKline, who invented paroxetine. So it's no surprise that the text paints GSK and their product in a favourable light, but the data warrant a close look and the results are rather interesting - and complicated.
They took all of the placebo-controlled trials on paroxetine for any psychiatric disorder - because it wasn't just trialled in depression, but also in PTSD, anxiety, and more. They excluded studies with fewer than 30 people; this makes sense though it's somewhat arbitrary, why not 40 or 20? Anyway, they ended up with 61 trials.
First they looked at suicide. In a nutshell paroxetine increased suicidal "behaviour or ideation" in younger patients (age 25 or below) relative to placebo, whether or not they were being treated for depression. In older patients, it only increased suicidality in the depression trials, and the effect was smaller. I've put a red dot where paroxetine was worse than placebo; this doesn't mean the effect was "statistically significant", but the numbers are so small that this is fairly meaningless. Just look at the numbers. This is not very new. It's been accepted for a while that broadly the same applies when you look at trials of other antidepressants. Whether this causes extra suicides in the real world is a big question.
This is not very new. It's been accepted for a while that broadly the same applies when you look at trials of other antidepressants. Whether this causes extra suicides in the real world is a big question.
When it comes to efficacy, however, we find some rather startling info that's not been presented together in one article before, to my knowledge. Here's a graph showing the effect of paroxetine over-and-above placebo in all the different disorders, expressed as a proportion of the improvement seen in the placebo group. Now I should point out that I just made this measure up. It's not ideal. If the placebo response is very small, then a tiny drug effect will seem large by comparison, even if what this really means is that neither drug nor placebo do any good.
Now I should point out that I just made this measure up. It's not ideal. If the placebo response is very small, then a tiny drug effect will seem large by comparison, even if what this really means is that neither drug nor placebo do any good.
However the flip side of that coin is that it controls for the fact that rating scales for different disorders might be just more likely to show change than others. The d score is a more widely used standardized measure of effect size - though it has its own shortcomings - and I'd like to know those, but the data they provide don't allow us to easily calculate it. You could do it from the GSK database but it would take ages.
Anyway as you can see paroxetine was better, relative to placebo, against PTSD, PMDD, obsessive-compulsive disorder, and social anxiety, than it was against depression measured with the "gold-standard" HAMD scale! In fact the only thing it was worse against was Generalized Anxiety Disorder. Using the alternative MADRS depression scale, the antidepressant effect was bigger, but still small compared to OCD and social anxiety.
This is rather remarkable. Everyone calls paroxetine "an antidepressant", yet at least in one important sense it works better against OCD and social anxiety than it does against depression!
In fact, is paroxetine an antidepressant at all? It works better on MADRS and very poorly on the HAMD; is this because the HAMD is a better scale of depression, and the MADRS actually measures anxiety or OCD symptoms?
That's a lovely neat theory... but in fact the HAMD-17 has two questions about anxiety, scoring 0-4 points each, so you can score up to 8 (or 12 if you count "hypochondriasis", which is basically health anxiety, so you probably should), out of a total maximum of 52. The MADRS has one anxiety item with a max score of 6 on a total of 60. So the HAMD is more "anxious" than the MADRS.
This is more than just a curiosity. Paroxetine's antidepressant effect was tiny in those aged 25 or under on the HAMD - treatment just 9% of the placebo effect - but on the MADRS in the same age group, the benefit was 35%! So what is the HAMD measuring and why is it different to the MADRS?
Honestly, it's hard to tell because the Hamilton scale is so messy. It measures depression and the other distressing symptoms which commonly go along with it. The idea, I think, was that it was meant to be a scale of the patient's overall clinical severity - how seriously they were suffering - rather than a measure of depression per se.
Which is fine. Except that most modern trials carefully exclude anyone with "comorbid" symptoms like anxiety, and on the other hand, recruit people with symptoms quite different to the depressed inpatients that Dr Max Hamilton would have seen when he invented the scale in 1960.
Yet 50 years later the HAMD17, unmodified, is still the standard scale. It's been repeatedly shown to be multi-factorial (it doesn't measure one thing), no-one even agrees on how to interpret it, and a "new scale", the HAMD6, which consists of simply chucking out 11 questions and keeping the 6 that actually measure depression, has been shown to be better. Yet everyone still uses the HAMD17 because everyone else does.
Link: I recently covered a dodgy paper about paroxetine in adolescents with depression; it wasn't included in this analysis because this was about adults.
Paxil: The Whole Truth?
 03.20
03.20
 wsn
wsn
 Posted in
antidepressants,
drugs,
mental health,
papers,
placebo
Posted in
antidepressants,
drugs,
mental health,
papers,
placebo
The Mystery of "Whoonga"
06.05
wsn

According to a disturbing BBC news story, South African drug addicts are stealing medication from HIV+ people and using it to get high:
'Whoonga' threat to South African HIV patients"Whoonga" is, allegedly, the street name for efavirenz (aka Stocrin), one of the most popular antiretroviral drugs. The pills are apparantly crushed, mixed with marijuana, and smoked for its hallucinogenic effects.
This is not, in fact, a new story; Scientific American covered it 18 months ago and the BBC themselves did in 2008 (although they didn't name efavirenz.)
Edit 16.00 pm: In fact the picture is even messier than I first thought. Some sources, e.g. Wikipedia and the articles it links to, mostly from South Africa, suggest that "whoonga" is actually a 'brand' of heroin and that the antiretrovirals may not be the main ingredient, if they're an ingredient at all. If this is true, then the BBC article is misleading. Edit and see the Comments for more on this...
Why would an antiviral drug get you high? This is where things get rather mysterious. Efavirenz is known to enter the brain, unlike most other HIV drugs, and psychiatric side-effects including anxiety, depression, altered dreams, and even hallucinations are common in efavirenz use, especially with high doses (1,2,3), but they're usually mild and temporary. But what's the mechanism?
No-one knows, basically. Blank et al found that efavirenz causes a positive result on urine screening for benzodiazepines (like Valium). This makes sense given the chemical structure:
 Efavirenz is not a benzodiazepine, because it doesn't have the defining diazepine ring (the one with two Ns). However, as you can see, it has a lot in common with certain benzos such as oxazepam and lorazepam.
Efavirenz is not a benzodiazepine, because it doesn't have the defining diazepine ring (the one with two Ns). However, as you can see, it has a lot in common with certain benzos such as oxazepam and lorazepam.However, while this might well explain why it confuses urine tests, it doesn't by itself go far to explaining the reported psychoactive effects. Oxazepam and lorazepam don't cause hallucinations or psychosis, and they reduce anxiety, rather than causing it.
They also found that efavirenz caused a false positive for THC, the active ingredient in marijuana; this was probably caused by the gluconuride metabolite. Could this metabolite have marijuana-like effects? No-one knows at present.
Beyond that there's been little research on the effects of efavirenz in the brain. This 2010 paper reviewed the literature and found almost nothing. There were some suggestions that it might affect inflammatory cytokines or creatine kinase, but these are not obvious candidates for the reported effects.
Could the liver be responsible, rather than the brain? Interestingly, the 2010 paper says that efavirenz inhibits three liver enzymes: CYPs 2C9, 2C19, and 3A4. All three are involved in the breakdown of THC, so, in theory, efavirenz might boost the effects of marijauna by this mechanism - but that wouldn't explain the psychiatric side effects seen in people who are taking the drug for HIV and don't smoke weed.
Drugs that cause hallucinations generally either agonize 5HT2A receptors or block NMDA receptors. Off the top of my head, I can't see any similarities between efavirenz and drugs that target those systems like LCD (5HT2A) or ketamine or PCP (NMDA), but I'm no chemist and anyway, structural similarity is not always a good guide to what drugs do.
If I were interested in working out what's going on with efavirenz, I'd start by looking at GABA, the neurotransmitter that's the target of benzos. Maybe the almost-a-benzodiazepine-but-not-quite structure means that it causes some unusual effects on GABA receptors? No-one knows at present. Then I'd move on to 5HT2A and NMDA receptors.
Finally, it's always possible that the users are just getting stoned on cannabis and mistakenly thinking that the efavirenz is making it better through the placebo effect. Stranger things have happened. If so, it would make the whole situation even more tragic than it already is.
Cavalcante GI, Capistrano VL, Cavalcante FS, Vasconcelos SM, Macêdo DS, Sousa FC, Woods DJ, & Fonteles MM (2010). Implications of efavirenz for neuropsychiatry: a review. The International journal of neuroscience, 120 (12), 739-45 PMID: 20964556 Posted in
drugs,
media,
mental health
The Mystery of Stiff Person Syndrome
11.30
wsn
"Stiff Person Syndrome" (SPS) is a rare neurological disease with a silly name but serious symptoms. Not in fact a disorder caused by an overdose of Viagra, the defining feature of SPS is uncontrollable muscle rigidity, which comes and goes in bouts, but generally gets worse over time. However, other symptoms are seen including depression, anxiety, and other neurological features such as cerebellar ataxia.
Not in fact a disorder caused by an overdose of Viagra, the defining feature of SPS is uncontrollable muscle rigidity, which comes and goes in bouts, but generally gets worse over time. However, other symptoms are seen including depression, anxiety, and other neurological features such as cerebellar ataxia.
What causes SPS? Well, it's been known for over 20 years that most SPS patients have antibodies against the enzyme GAD65, which is required for the production of GABA, the main inhibitory neurotransmitter in the brain. The body shouldn't be producing antibodies against its own proteins, but unfortunately this does happen quite often, for various reasons, and the result is autoimmune diseases.
So this all seems to make sense. We know that GABA causes muscle relaxation by reducing the brain's input to the muscles. This is why GABA drugs like Valium are muscle-relaxants, and it's part of the reason why drunk people tend to stagger around.
This also explains the anxiety symptoms, because Valium and beer make you less anxious, while drugs that block GABA cause panic attacks. Anti-GAD65 antibodies block GAD, so less GABA gets made. So SPS is autoimmunity against GAD65. Mystery solved?
Not quite. Anti-GAD65 antibodies are also seen in most people with Type I diabetes, but the vast majority of diabetics luckily don't suffer SPS. Mystery remains.
Two studies just out investigated exactly what the antibodies produced by SPS patients do. Geis et al purified the antibodies from a 53 year old woman with SPS and serious anxiety, and injected them into the brains of some rats.
The rats became very anxious. Here's what the cowardly critters did in a standard rodent anxiety test: they avoided the open spaces, which are naturally scary to rodents, who prefer dark, enclosed places: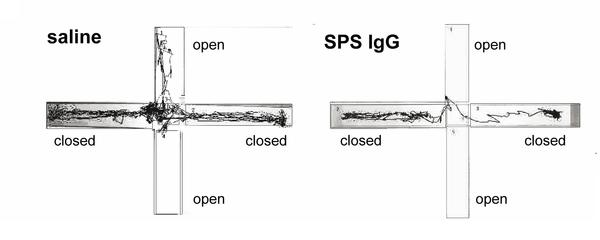 This was associated with reduced GABA production.
This was associated with reduced GABA production.
Meanwhile Manto et al found that anti-GAD65 antibodies from another patient with SPS caused very different effects in rat brains compared to the antibodies derived from a patient with autoimmune cerebellar ataxia, but no SPS symptoms. They also found that two kinds of off-the-shelf anti-GAD65 antibodies commonly used in research had different effects as well.
Taken together this all suggests that SPS is caused by anti-GAD65 antibodies, but they have to be a particular type. Different antibodies cause different symptoms even though they all bind to GAD65.
Presumably this is because it's a big protein, and antibodies could bind to any part of it. Only ones that block the "business end" - the part which actually catalyzes the formation of GABA - will cause problems. A bit like how if you get shot in the heart, that's the end of you, but get shot in the foot and it probably won't be.
Manto MU, Hampe CS, Rogemond V, & Honnorat J (2011). Respective implications of glutamate decarboxylase antibodies in stiff person syndrome and cerebellar ataxia. Orphanet journal of rare diseases, 6 (1) PMID: 21294897
Posted in
animals,
drugs,
mental health,
papers
Antidepressants Don't Work...In Fish
13.10
wsn
Here at Neuroskeptic fMRI scanning and antidepressants are both big topics.
As I discussed lask week, fish - specifically salmon - are the next big thing in fMRI and the number of salmon brains being scanned is growing at a remarkable rate. But fish haven't made much of an entrance into the world of antidepressants...until now.
Swedish scientists Holmberg et al have just published a paper asking: Does waterborne citalopram affect the aggressive and sexual behaviour of rainbow trout and guppy?
SSRI antidepressants, of which citalopram is one, are very popular. So popular, in fact, that non-trivial levels of SSRIs have been found in sewage and there's a concern that they might make their way into lakes and rivers and thereby affect the behaviour of the animals living there.
Holmberg et al set out to see what citalopram did to some fish in an attempt to find out whether this is likely to be a major problem. So they put some citalopram in the fish's water supplies and then tested their aggressiveness and also their sex drives. It turns out that one of the main ways of measure fish aggression is to put a mirror in their tank and see if they try to fight their own reflection. Fish are not very bright, really.
Anyway, the good news for fish everywhere was that seven days of citalopram exposure had no effect at all, even at doses much higher than those reported as a pollutant (the maximum dose was 0.1 mg/l). And the authors had no conflicts of interest: Big Pharma had nothing to do with this research, although Big Fish Farmer did because they bought the fish from one.
However, this may not be the end of the story, because it turned out that citalopram was very poorly absorbed into the fish's bloodstreams. But other antidepressants have been reported to accumulate in fish. Clearly, the only way to find out for sure what's going on would be to use fMRI...
Posted in
animals,
antidepressants,
drugs,
fMRI,
funny,
mental health,
papers
Retract That Seroxat?
00.45
wsn
Should a dodgy paper on antidepressants be retracted? And what's scientific retraction for, anyway?
Read all about it in a new article in the BMJ: Rules of Retraction. It's about the efforts of two academics, Jon Jureidini and Leemon McHenry. Their mission - so far unsuccesful - is to get this 2001 paper retracted: Efficacy of paroxetine in the treatment of adolescent major depression.
Jureidini is a member of Healthy Skepticism, a fantastic Australian organization that Neuroskeptic readers have encountered before. They've got lots of detail on the ill-fated "Study 329", including internal drug company documents, here.
So what's the story? Study 329 was a placebo-controlled trial of the SSRI paroxetine (Paxil, Seroxat) in 275 depressed adolescents. The paper concluded: that "Paroxetine is generally well tolerated and effective for major depression in adolescents." It was published in the Journal of the American Academy of Child and Adolescent Psychiatry (JAACAP).
There's two issues here: whether paroxetine worked, and whether it was safe. On safety, the paper concluded that "Paroxetine was generally well tolerated...and most adverse effects were not serious." Technically true, but only because there were so many mild side effects.
In fact, 11 patients on paroxetine reported serious adverse events, including suicidal ideation or behaviour, and 7 were hospitalized. Just 2 patients in the placebo group had such events. Yet we are reassured that "Of the 11, only headache (1 patient) was considered by the treating investigator to be related to paroxetine treatment."
The drug company argue that it didn't become clear that paroxetine caused suicidal ideation in adolescents until after the paper was published. In 2002, British authorities reviewed the evidence and said that paroxetine should not be given in this age group.
That's as maybe; the fact remains that in this paper there was a strongly raised risk. However, in fairness, all that data was there in the paper, for readers to draw their own conclusions from. The paper downplays it, but the numbers are there.
*
The efficacy question is where the allegations of dodgy practices are most convincing. The paper concludes that paroxetine worked, while imipramine, an older antidepressant, didn't.
Jureidini and McHenry say that paroxetine only worked on a few of the outcomes - ways of measuring depression and how much the patients improved. On most of the outcomes, it didn't work, but the paper focusses on the ones where it did. According to the BMJ
Here's the worst example. In the original protocol, two "primary" endpoints were specified: the change in the total Hamilton Scale (HAMD) score, and % of patients who 'responded', defined as either an improvement of more than 50% of their starting HAMD score or a final HAMD of 8 or below.Study 329’s results showed that paroxetine was no more effective than the placebo according to measurements of eight outcomes specified by Martin Keller, professor of psychiatry at Brown University, when he first drew up the trial.
Two of these were primary outcomes...the drug also showed no significant effect for the initial six secondary outcome measures. [it] only produced a positive result when four new secondary outcome measures, which were introduced following the initial data analysis, were used... Fifteen other new secondary outcome measures failed to throw up positive results.
On neither of these measures did paroxetine work better than placebo at the p=0.05 significance level. It did work if you defined 'responded' to mean only a final HAMD of 8 or below, but this was not how it was defined in the protocol. In fact, the Methods section of the paper follows the protocol faithfully. Yet in the Results section, the authors still say that:
Of the depression-related variables, paroxetine separated statistically from placebo at endpoint among four of the parameters: response (i.e., primary outcome measure)...It may seem like a subtle point. But it's absolutely crucial. Paroxetine just did not work on either pre-defined primary outcome measure, and the paper says that it did.
Finally, there were also issues of ghostwriting. I've never been that concerned by this in itself. If the science is bad, it's bad whoever wrote it. Still, it's hardly a good thing.
Does any of this matter? In one sense, no. Authorities have told doctors not to use paroxetine in adolescents with depression since 2002 (in the UK) and 2003 (in the USA). So retracting this paper wouldn't change much in the real world of treatment.
But in another sense, the stakes are enormous. If this paper were retracted, it would set a precedent and send a message: this kind of p-value fishing to get positive results, is grounds for retraction.
This would be huge, because this kind of fishing is sadly very common. Retracting this paper would be saying: selective outcome reporting is a form of misconduct. So this debate is really not about Seroxat, but about science.

There are no Senates or Supreme Courts in science. However, journal editors are in a unique position to help change this. They're just about the only people (grant awarders being the others) who have the power to actually impose sanctions on scientists. They have no official power. But they have clout.
Were the JAACAP to retract this paper, which they've so far said they have no plans to do, it would go some way to making these practices unacceptable. And I think no-one can seriously disagree that they should be unacceptable, and that science and medicine would be much better off if they were. Do we want more papers like this, or do we want fewer?
So I think the question of whether to retract or not boils down to whether it's OK to punish some people "to make an example of them", even though we know of plenty of others who have done the same, or worse, and won't be punished.
My feeling is: no, it's not very fair, but we're talking about multi-billion pound companies and a list of authors whose high-flying careers are not going to crash and burn just because one paper from 10 years ago gets pulled. If this were some poor 24 year old's PhD thesis, it would be different, but these are grown-ups who can handle themselves.
So I say: retract.
Newman, M. (2010). The rules of retraction BMJ, 341 (dec07 4) DOI: 10.1136/bmj.c6985Keller MB, et al. (2001). Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled trial. Journal of the American Academy of Child and Adolescent Psychiatry, 40 (7), 762-72 PMID: 11437014
Posted in
antidepressants,
bad neuroscience,
drugs,
mental health,
papers
Depression Treatment Increased From 1998 to 2007
07.50
wsn
A paper just out reports on the changing patterns of treatment for depression in the USA, over the period from 1998 to 2007. The headline news is that it increased: the overall rate of people treated for some form of "depression" went from 2.37% to 2.88% per year. That's an increase of 21%, which is not trivial, but it's much less than the increase in the previous decade: it was just 0.73% in 1987.
The headline news is that it increased: the overall rate of people treated for some form of "depression" went from 2.37% to 2.88% per year. That's an increase of 21%, which is not trivial, but it's much less than the increase in the previous decade: it was just 0.73% in 1987.
But the increase was concentrated in. some groups of people.
- Americans over 50 accounted for the bulk of the rise. Their use went up by about 50%, while rates in younger people stayed almost steady. In '98 the peak age band was 35-49, now it's 50-64, with almost 5% of those people getting treated in any given year.
- Men's rates of treatment went up by over 40% while women's only increased by 10%. Women are still more likely to get treated for depression than men, though, with a ratio of 1.7 women for each 1 man. But that ratio is a lot closer than it used to be.
- Black people's rates increased hugely, by 120%. Rates in black people now stand at 2.2% which is close behind whites at 3.2%. Hispanics are now the least treated major ethnic group at 1.9%: in previous studies, blacks were the least treated. (There was no data on Asians or others).
In terms of what treatments people got, out of everyone treated for depression, 80% got some kind of drugs, and that didn't change much. But use of psychotherapy declined a bit from 54% to 43% (some people got both).
What's also interesting is that the same authors reported last year that, over pretty much the same time period ('96 to '05), the number of Americans who used antidepressants in any given year sky-rocketed from 5% to 10% - that is to say, much faster than the rate of depression treatment rose! And the data are comparable, because they came from the same national MEPS surveys.
In other words, the decade must have seen antidepressants increasingly being used to treat stuff other than depression. What stuff? Well, all kinds of things. SSRIs are popular in everything from anxiety and OCD to premature ejaculation. Several of the "other new" drugs, like mirtazapine and trazodone, are very good at putting you to sleep (rather too good, some users would say...)
Marcus SC, & Olfson M (2010). National trends in the treatment for depression from 1998 to 2007. Archives of general psychiatry, 67 (12), 1265-73 PMID: 21135326 Posted in
1in4,
antidepressants,
drugs,
mental health,
papers
The Town That Went Mad
06.30
wsn
Pont St. Esprit is a small town in southern France. In 1951 it became famous as the site of one of the most mysterious medical outbreaks of modern times. As Dr's Gabbai, Lisbonne and Pourquier wrote to the British Medical Journal, 15 days after the "incident":
As Dr's Gabbai, Lisbonne and Pourquier wrote to the British Medical Journal, 15 days after the "incident":
The first symptoms appeared after a latent period of 6 to 48 hours. In this first phase, the symptoms were generalized, and consisted in a depressive state with anguish and slight agitation.In most patients, these symptoms, including the total insomnia, persisted for several days. In some of the patients, these symptoms progressed to full-blown psychosis:
After some hours the symptoms became more clearly defined, and most of the patients presented with digestive disturbances... Disturbances of the autonomic nervous system accompanied the digestive disorders-gusts of warmth, followed by the impression of "cold waves", with intense sweating crises. We also noted frequent excessive salivation.
The patients were pale and often showed a regular bradycardia (40 to 50 beats a minute), with weakness of the pulse. The heart sounds were rather muffled; the extremities were cold... Thereafter a constant symptom appeared - insomnia lasting several days... A state of giddiness persisted, accompanied by abundant sweating and a disagreeable odour. The special odour struck the patient and his attendants.
Logorrhoea [speaking a lot], psychomotor agitation, and absolute insomnia always presaged the appearance of mental disorders. Towards evening visual hallucinations appeared, recalling those of alcoholism. The particular themes were visions of animals and of flames. All these visions were fleeting and variable.
In many of the patients they were followed by dreamy delirium. The delirium seemed to be systematized, with animal hallucinations and self-accusation, and it was sometimes mystical or macabre. In some cases terrifying visions were followed by fugues, and two patients even threw themselves out of windows... Every attempt at restraint increased the agitation.
In severe cases muscular spasms appeared, recalling those of tetanus, but seeming to be less sustained and less painful... The duration of these periods of delirium was very varied. They lasted several hours in some patients, in others they still persist.
At first, the cause was assumed to be ergotism - poisoning caused by chemicals produced by a fungus which can infect grain crops. Contaminated bread was, therefore, thought to be responsible. Ergotism produces symptoms similar to those reported at Pont St. Esprit, including hallucinations, because some of the toxins are chemically related to LSD.
However, there have been other theories. Some (including Albert Hofmann, the inventor of LSD) attribute the poisoning to pesticides containing mercury, or to the flour bleaching agent nitrogen trichloride.
More recently, journalist Hank Albarelli claimed that it was in fact a CIA experiment to test out the effects of LSD as a chemical weapon, though this is disputed. What really happened is, in other words, still a mystery.
Link: The Crazies (2010) is a movie about a remarkably similar outbreak of mass insanity in a small town.
GABBAI, LISBONNE, & POURQUIER (1951). Ergot poisoning at Pont St. Esprit. British medical journal, 2 (4732), 650-1 PMID: 14869677 Posted in
drugs,
history,
media,
mental health,
papers
Worst. Antidepressant. Ever.
04.22
wsn
Reboxetine is an antidepressant. Except it's not, because it doesn't treat depression.
 This is the conclusion of a much-publicized article just out in the BMJ: Reboxetine for acute treatment of major depression: systematic review and meta-analysis of published and unpublished placebo and SSRI controlled trials.
This is the conclusion of a much-publicized article just out in the BMJ: Reboxetine for acute treatment of major depression: systematic review and meta-analysis of published and unpublished placebo and SSRI controlled trials.
Reboxetine was introduced to some fanfare, because its mechanism of action is unique - it's a selective norepinephrine reuptake inhibitor (NRI), which has no effect on serotonin, unlike Prozac and other newer antidepressants. Several older tricyclic antidepressants were NRIs, but they weren't selective because they also blocked a shed-load of receptors.
So in theory reboxetine treats depression while avoiding the side effects of other drugs, but last year, Cipriani et al in a headline-grabbing meta-analysis concluded that in fact it's the exact opposite: reboxetine was the least effective new antidepressant, and was also one of the worst in terms of side effects. Oh dear.
And that was only based on the published data. It turns out that Pfizer, the manufacturers of reboxetine, had chosen to not publish the results of most of their clinical trials of the drug, because the data showed that it was crap.
The new BMJ paper includes these unpublished results - it took an inordinate amount of time and pressure to make Pfizer agree to share them, but they eventually did - and we learn that reboxetine is:
- no more effective than a placebo at treating depression.
- less effective than SSRIs, which incidentally are better than placebo in this dataset (a bit).
- worse tolerated than most SSRIs, and much worse tolerated than placebo.
 Claims that reboxetine is dangerous on the basis of this study are a bit misleading - it may be, but there was no evidence for that in these data. It caused nasty and annoying side-effects, but that's not the same thing, because if you don't like side-effects, you could just stop taking it (which is what many people in these trials did).
Claims that reboxetine is dangerous on the basis of this study are a bit misleading - it may be, but there was no evidence for that in these data. It caused nasty and annoying side-effects, but that's not the same thing, because if you don't like side-effects, you could just stop taking it (which is what many people in these trials did).Anyway, what are the lessons of this sorry tale, beyond reboxetine being rubbish? The main one is: we have to start forcing drug companies and other researchers to publish the results of clinical trials, whatever the results are. I've discussed this previously and suggested one possible way of doing that.
The situation regarding publication bias is far better than it was 10 years ago, thanks to initiatives such as clinicaltrials.gov; almost all of the reboxetine trials were completed before the year 2000; if they were run today, it would have been much harder to hide them, but still not impossible, especially in Europe. We need to make it impossible, everywhere, now.
The other implication is, ironically, good news for antidepressants - well, except reboxetine. The existence of reboxetine, a drug which has lots of side effects, but doesn't work, is evidence against the theory (put forward by Joanna Moncrieff, Irving Kirsch and others) that even the antidepressants that do seem to work, only work because of active placebo effects driven by their side effects.
So given that reboxetine had more side effects than SSRIs, it ought to have worked better, but actually it worked worse. This is by no means the nail in the coffin of the active placebo hypothesis but it is, to my mind, quite convincing.
Link: This study also blogged by Good, Bad and Bogus.
Eyding, D., Lelgemann, M., Grouven, U., Harter, M., Kromp, M., Kaiser, T., Kerekes, M., Gerken, M., & Wieseler, B. (2010). Reboxetine for acute treatment of major depression: systematic review and meta-analysis of published and unpublished placebo and selective serotonin reuptake inhibitor controlled trials BMJ, 341 (oct12 1) DOI: 10.1136/bmj.c4737 Posted in
antidepressants,
bad neuroscience,
drugs,
mental health,
papers,
science
Cannabinoids in Huntington's Disease
05.10
wsn
Two recent papers have provided strong evidence that the brain's endocannabinoid system is dysfunctional in Huntington's Disease, paving the way to possible new treatments. Huntington's Disease is a genetic neurological disorder. Symptoms generally appear around age 40, and progress gradually from subtle movement abnormalities to dementia and complete loss of motor control. It's incurable, although medication can mask some of the symptoms. Singer Woodie Guthrie is perhaps the disease's best known victim: he ended his days in a mental institution.
Huntington's Disease is a genetic neurological disorder. Symptoms generally appear around age 40, and progress gradually from subtle movement abnormalities to dementia and complete loss of motor control. It's incurable, although medication can mask some of the symptoms. Singer Woodie Guthrie is perhaps the disease's best known victim: he ended his days in a mental institution.
The biology of Huntington's is only partially understood. It's caused by mutations in the huntingtin gene, which lead to the build-up of damaging proteins in brain cells, especially in the striatum. But exactly how this produces symptoms is unclear.
The two new papers show that cannabinoids play an important role. First off, Van Laere et al used PET imaging to measure levels of CB1 receptors in the brain of patients in various stages of Huntington's. CB1 is the main cannabinoid receptor in the brain; it responds to natural endocannabinoid neurotransmitters, and also to THC, the active ingredient in marijuana.
They found serious reductions in all areas of the brain compared to healthy people, and interestingly, the loss of CB1 receptors occurred early in the course of the disease: That was an important finding, but it didn't prove that CB1 loss was causing any problems: it might have just been a side-effect of the disease. Now another study using animals has shown that it's not: Blazquez et al. They studied mice with the same mutation that causes Huntington's in humans. These unfortunate rodents develop Huntington's, unsurprisingly.
That was an important finding, but it didn't prove that CB1 loss was causing any problems: it might have just been a side-effect of the disease. Now another study using animals has shown that it's not: Blazquez et al. They studied mice with the same mutation that causes Huntington's in humans. These unfortunate rodents develop Huntington's, unsurprisingly.
They found that Huntington's mice who also had a mutation eliminating the CB1 receptor suffered more severe symptoms, which appeared earlier, and progressed faster. This suggests that CB1 plays a neuroprotective role, which is consistent with lots of earlier studies in other disorders.
If so, drugs that activate CB1 - like THC - might be able to slow down the progression of the disease, and indeed it did: Huntington's mice given THC injections stayed healthier for longer, although they eventually succumbed to the disease. Further experiments showed that mutant huntingtin switches off expression of the CB1 receptor gene, explaining the loss of CB1.
This graph shows performance on the RotaRod test of co-ordination: mice with Huntington's (R6/2) got worse and worse starting at 6 weeks of age (white bars), but THC slowed down the decline (black bars). The story was similar for other symptoms, and for the neural damage seen in the disease. They conclude that:
They conclude that:
Altogether, these results support the notion that downregulation of type 1 cannabinoid receptors is a key pathogenic event in Huntington’s disease, and suggest that activation of these receptors in patients with Huntington’s disease may attenuate disease progression.Now, this doesn't mean people with Huntington's should be heading out to buy Bob Marley posters and bongs just yet. For one thing, Huntington's disease often causes psychiatric symptoms, including depression and psychosis. Cannabis use has been linked to psychosis fairly convincingly, so marijuana might make those symptoms worse.
Still, it's very promising. In particular, it will be interesting to try out next-generation endocannabinoid boosting drugs, such as FAAH inhibitors, which block the breakdown of anandamide, one of the most important endocannabinoids.
In animals FAAH inhibitors have pain relieving, anti-anxiety, and other beneficial effects, but they don't cause the same behavioural disruptions that THC does. This suggests that they wouldn't get people high, either, but there's no published data on what they do in humans yet...
Van Laere K, et al. (2010). Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. Journal of nuclear medicine : official publication, Society of Nuclear Medicine, 51 (9), 1413-7 PMID: 20720046Blázquez C, et al. (2010). Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain : a journal of neurology PMID: 20929960
Posted in
animals,
CNR1,
drugs,
mental health,
papers
Stopping Antidepressants: Not So Fast
08.00
wsn
People who quit antidepressants slowly, by gradually decreasing the dose, are much less likely to suffer a relapse, according to Baldessarini et al. in the American Journal of Psychiatry. They describe a large sample (400) of patients from Sardinia, Italy, who had responded well to antidepressants, and then stopped taking them. The antidepressants had been prescribed for either depression, or panic attacks.
They describe a large sample (400) of patients from Sardinia, Italy, who had responded well to antidepressants, and then stopped taking them. The antidepressants had been prescribed for either depression, or panic attacks.
People who quit suddenly (over 1-7 days) were more likely to relapse, and relapsed sooner, than the ones who stopped gradually (over a period of 2 weeks or more). This graph shows what % of the patients in each group remained well at each time point (in terms of days since their final pill.) As you can see, the two lines separate early, and then remain apart by about the same distance (20%) for the whole 12 months.
This graph shows what % of the patients in each group remained well at each time point (in terms of days since their final pill.) As you can see, the two lines separate early, and then remain apart by about the same distance (20%) for the whole 12 months.
What this means is that rapid discontinuation didn't just accelerate relapses that were "going to happen anyway". It actually caused more relapses - about 1 in 5 "extra" people. These "extra" relapses all happened in the first 3 months, because after that, the slope of the lines is identical.
On the other hand, they rarely happened immediately - it's not as if people relapsed within days of their last pill. The pattern was broadly similar for older antidepressants (tricyclics) and newer ones (SSRIs).
The authors note that these data throw up important questions about "relapse prevention" trials comparing people who stay on antidepressants vs. those who are switched - abruptly - to placebo. People who stay on the drug usually do better, but is this because the drug works, or because the people on placebo were withdrawn too fast?
This was an observational study, not an experiment. There was no randomization. People quit antidepressants for various "personal or clinical reasons"; 80% of the time it was their own decision, and only 20% of the time was it due to their doctor's advice.
So it's possible that there was some underlying difference between the two groups, that could explain the differences. Regression analysis revealed that the results weren't due to differences in dose, duration of treatment, diagnosis, age etc., but you can't measure every possible confound.
Only randomized controlled trials could provide a final answer, but there's little chance of anyone doing one. Drug companies are unlikely to fund a study about how to stop using their products. So we have only observational data to go on. These data fit in with previous studies showing that there's a similar story when it comes to quitting lithium and antipsychotics. Gradual is better.
But that's common sense. Tapering medications slowly is a good idea in general, because it gives your system more time to adapt. Of course, sometimes there are overriding medical reasons to quit quickly, but apart from in such cases, I'd always want to come off anything as gradually as possible.
Posted in
antidepressants,
drugs,
mental health,
papers
Shotgun Psychiatry
07.50
wsn
There's a paradox at the heart of modern psychiatry, according to an important new paper by Dr Charles E. Dean, Psychopharmacology: A house divided. It's a long and slightly rambling article, but Dean's central point is pretty simple. The medical/biological model of psychiatry assumes that there are such things as psychiatric diseases. Something biological goes wrong, presumably in the brain, and this causes certain symptoms. Different pathologies cause different symptoms - in other words, there is specificity in the relationship between brain dysfunction and mental illness.
It's a long and slightly rambling article, but Dean's central point is pretty simple. The medical/biological model of psychiatry assumes that there are such things as psychiatric diseases. Something biological goes wrong, presumably in the brain, and this causes certain symptoms. Different pathologies cause different symptoms - in other words, there is specificity in the relationship between brain dysfunction and mental illness.
Psychiatric diagnosis rests on this assumption. If and only if we can use a given patient's symptoms to infer what kind of underlying illness they have (schizophrenia, bipolar disorder, depression), diagnosis makes sense. This is why we have DSM-IV which consists of a long list of disorders, and the symptoms they cause. Soon we'll have DSM-V.
The medical model has been criticized and defended at great length, but Dean doesn't do either. He simply notes that modern psychiatry has in practice mostly abandoned the medical model, and the irony is, it's done this because of medicines.
If there are distinct psychiatric disorders, there ought to be drugs that treat them specifically. So if depression is a brain disease, say, and schizophrenia is another, there ought to be drugs that only work on depression, and have no effect on schizophrenia (or even make it worse.) And vice versa.
But, increasingly, psychiatric drugs are being prescribed for multiple different disorders. Antidepressants are used in depression, but also all kinds of anxiety disorders (panic, social anxiety, general anxiety), obsessive-compulsive disorder, PTSD, and more. Antipsychotics are also used in mania and hypomania, in kids with behaviour problems, and increasingly in depression, leading some to complain that the term "antipsychotics" is misleading. And so on.
So, Dean argues, in clinical practice, psychiatrists don't respect the medical model - yet that model is their theoretical justification for using psychiatric drugs in the first place.
He looks in detail at one particularly curious case: the use of atypical antipsychotics in depression. Atypicals, like quetiapine (Seroquel) and olanzapine (Zyprexa), were originally developed to treat schizophrenia and other psychotic states. They are reasonably effective, though most of them are no more so than older "typical" antipsychotics.
Recently, atypicals have become very popular for other indications, most of all mood disorders: mania and depression. Their use in mania is perhaps not so surprising, because severe mania has much in common with psychosis. Their use in depression, however, throws up many paradoxes (above and beyond how one drug could treat both mania and its exact opposite, depression.)
Antipsychotics block dopamine D2 receptors. Psychosis is generally considered to be a disorder of "too much dopamine", so that makes sense. The dopamine hypothesis of psychosis and antipsychotic action is 50 years old, and still the best explanation going.
But depression is widely considered to involve too little dopamine, and there is lots of evidence that almost all antidepressants (indirectly) increase dopamine release. Wouldn't that mean that antidepressants could cause psychosis (they don't?). And why, Dean asks, would atypicals, that block dopamine, help treat depression?
Maybe it's because they also act on other systems? On top of being D2 antagonists, atypicals are also serotonin 5HT2A/C receptor blockers. Long-term use of antidepressants reduces 5HT2 levels, and some antidepressants are also 5HT2 antagonists, so this fits. However, it creates a paradox for the many people who believe that 5HT2 antagonism is important for the antipsychotic effect of atypicals as well - if that were true, antidepressants should be antipsychotics as well (they're not.) And so on.
There may be perfectly sensible answers. Maybe atypicals treat depression by some mechanism that we don't understand yet, a mechanism which is not inconsistent with their also treating psychosis. The point is that there are many such questions standing in need of answers, yet psychopharmacologists almost never address them. Dean concludes:
it seems increasingly obvious that clinicians are actually operating from a dimensional paradigm, and not from the classic paradigm based on specificity of disease or drug... the disjunction between those paradigms and our approach to treatment needs to be recognized and investigated... Bench scientists need to be more familiar with current clinical studies, and stop using outmoded clinical research as a basis for drawing conclusions about the relevance of neurochemical processes to drug efficacy. Bench and clinical scientists need to fully address the question of whether the molecular/cellular/anatomical findings, even if interesting and novel, have anything to do with clinical outcome.
Dean CE (2010). Psychopharmacology: A house divided. Progress in neuro-psychopharmacology & biological psychiatry PMID: 20828593 Posted in
antidepressants,
bad neuroscience,
dbs,
drugs,
mental health,
papers,
schizophrenia
Are "Antipsychotics" Antipsychotics?
08.12
wsn
This is the question asked by Tilman Steinert & Martin Jandl in a letter to the journal Psychopharmacology. They point out that in the past 20 years, the word "antipsychotic" has exploded in popularity. Less than 100 academic papers were published with that word in the title in 1990, but now it's over 600 per year.
They point out that in the past 20 years, the word "antipsychotic" has exploded in popularity. Less than 100 academic papers were published with that word in the title in 1990, but now it's over 600 per year.
The older term for the same drugs was "neuroleptics". This terminology, however, has slowly but surely fallen into disuse over the same time period.
To illustrate this they have a nice graph of PubMed hits. Neuroskeptic readers will be familiar with these as I have often posted my own and I recently wrote a bash script to harvest this data automatically. Now you too can be a historian of medicine from the comfort of your own home...
Why does it matter what we call them? A name is just a name, right? No, that's the problem. Actually, neuroleptic is just a name, because it doesn't mean anything. The term derives from the Greek "neuron", meaning... neuron, and "lambanō" meaning "to take hold of". However, no-one knows that unless they look it up on Wikipedia because it's just a name.
Antipsychotic, on the other hand, means something: it means they treat psychosis. But whether or not this is an accurate description of what "antipsychotics" actually do, is controversial. For one thing, these drugs are also used to treat many non-psychotic illnesses, like depression, and PTSD.
More fundamentally, it's not universally accepted that they have a direct anti-psychotic effect. All antipsychotics are powerful sedatives. There's a school of thought that says that this is in fact all they are, and rather than treating psychosis, they just sedate people until they stop being obviously psychotic.
Personally, I don't believe that, but that's not really the point: the point is that it's controversial, and calling them antipsychotics makes it hard to think about that controversy in a sensible way. To say that antipsychotics aren't actually antipsychotic is a contradiction in terms. To say they are antipsychotic is a tautology. Names shouldn't dictate the terms of a debate in that way. A name should just be a name.
The same point applies to more than just antipsychotics - I mean neuroleptics - of course. Perhaps the worst example is "antidepressants". Prozac, for example, is called an antidepressant. Implying that it treats depression.
But according to clinical trials, Prozac and other SSRIs are a lot more effective, relative to placebo, in obsessive-compulsive disorders (OCD) than they are in depression (though this is not necessarily true of all "antidepressants", yet more evidence that the word is unhelpful.)
So, as I asked in a previous post: "Are SSRIs actually antiobsessives that happen to be helpful in some cases of depression?" Personally, I think the only name for them which doesn't make any questionable assumptions, is simply 'SSRIs'.
Posted in
drugs,
graphs,
mental health,
papers,
schizophrenia
Serotonin, Psychedelics and Depression
09.57
wsn
Note: This post is part of a Nature Blog Focus on hallucinogenic drugs in medicine and mental health, inspired by a recent Nature Reviews Neuroscience paper, The neurobiology of psychedelic drugs: implications for the treatment of mood disorders, by Franz Vollenweider & Michael Kometer. That article will be available, free (once you register), until September 23. For more information on this Blog Focus, see the "Table of Contents" here.
Neurophilosophy is covering the history of psychedelic psychiatry, while Mind Hacks provides a personal look at one particular drug, DMT. The Neurocritic discusses ketamine, an anesthetic with hallucinogenic properties, which is attracting a lot of interest at the moment as a treatment for depression. Ketamine, however, is not a "classical" psychedelic like the drugs that gave the 60s its unique flavor and left us with psychedelic rock, acid house and colorful artwork. Classical psychedelics are the focus of this post.
Ketamine, however, is not a "classical" psychedelic like the drugs that gave the 60s its unique flavor and left us with psychedelic rock, acid house and colorful artwork. Classical psychedelics are the focus of this post.
The best known are LSD ("acid"), mescaline, found in the peyote and a few other species of cactus, and psilocybin, from "magic" mushrooms of the Psilocybe genus. Yet there are literally hundreds of related compounds. Most of them are described in loving detail in the two heroic epics of psychopharmacology, PIKHaL and TIKHaL, written by chemists and trip veterans Alexander and Ann Shulgin.
The chemistry of psychedelics is closely linked with that of depression and antidepressants. All classical psychedelics are 5HT2A receptor agonists. Most of them have other effects on the brain as well, which contribute to the unique effects of each drug, but 5HT2A agonism is what they all have in common.
5HT2A receptors are excitatory receptors expressed throughout the brain, and are especially dense in the key pyramidal cells of the cerebral cortex. They're normally activated by serotonin (5HT), which is the neurotransmitter that's most often thought of as being implicated in depression. The relationship between 5HT and mood is very complicated, and depression isn't simply a disorder of "low serotonin", but there's strong evidence that it is involved.
There's one messy detail, which is that not quite all 5HT2A agonists are hallucinogenic. Lisuride, a drug used in Parkinson's disease, is closely related to LSD, and is a strong 5HT2A agonist, but it has no psychedelic effects. It's recently been shown that LSD and lisuride have different molecular effects on cortical cells, even though they act on the same receptor - in other words, there's more to 5HT2A than simply turning it "on" and "off".
How could psychedelics help to treat mental illness? On the face of it, the acute effects of these drugs - hallucinations, altered thought processes and emotions - sound rather like the symptoms of mental illness themselves, and indeed psychedelics have been referred to as "psychotomimetic" - mimicking psychosis.
There are two schools of thought here: psychological and neurobiological.
 The psychological approach ruled the first wave of psychedelic psychiatry, in the 50s and 60s. Psychiatry, especially in America, was dominated by Freudian theories of the unconscious. On this view, mental illness was a product of conflicts between unconscious desires and the conscious mind. The symptoms experienced by a particular patient were distressing, of course, but they also provided clues to the nature of their unconscious troubles.
The psychological approach ruled the first wave of psychedelic psychiatry, in the 50s and 60s. Psychiatry, especially in America, was dominated by Freudian theories of the unconscious. On this view, mental illness was a product of conflicts between unconscious desires and the conscious mind. The symptoms experienced by a particular patient were distressing, of course, but they also provided clues to the nature of their unconscious troubles.It was tempting to see the action of psychedelics as a weakening of the filters which kept the unconscious, unconscious - allowing repressed material to come into awareness. The only other time this happened, according to Freud, was during dreams. That's why Freud famously called the interpretation of dreams the "royal road to the unconscious".
Psychedelics offered analysts the tantalizing prospect of confronting the unconscious face-to-face, while awake, instead of having to rely on the patient's memory of their previous dreams. To enthusiastic Freudians, this promised to revolutionize therapy, in the same way that the x-ray had done so much for surgery. The "dreamlike" nature of many aspects of the psychedelic experience seemed to confirm this.
Not all psychedelic therapists were orthodox Freudians, however. There were plenty of other theories in circulation, many of them inspired by the theorists' own drug experiences. Stanislav Grof, Timothy Leary and others saw the psychedelic state of consciousness as the key to attaining spiritual, philosophical and even mystical insights, whether one was "ill" or "healthy" - and indeed, they often said that mental "illness" was itself a potential source of spiritual growth.
Like many things, psychiatry has changed since the 60s. Psychotherapy is currently dominated by cognitive-behavioural (CBT) theory, and Freudian ideas have gone distinctly out of fashion. It remains to be seen what CBT would make of LSD, but the basic idea - that carefully controlled use of drugs could help patients to "break through" psychological barriers to treatment - seems likely to remain at the heart of their continued use.
The other view is that these drugs could have direct biological effects which lead to improvements in mood. Repeated use of LSD, for example, has been shown to rapidly induce down-regulation of 5HT2A receptors. Presumably, this is the brain's way of "compensating" for prolonged 5HT2A activation. This is probably why tolerance to the effects of psychedelics rapidly develops, something that's long been known (and regretted) by heavy users.
Vollenweider and Kometeris note that this is interesting, because 5HT2A blockers are used as antidepressants - the drugs nefazadone and mirtazapine are the best known today, but most of the older tricyclic antidepressants are also 5HT2A antagonists. Atypical antipsychotics, which are also used in depression, are potent 5HT2A antagonists as well.
So indirectly suppressing 5HT2A might be one biological mechanism by which psychedelics improve mood. However, questions remain about how far this could explain any therapeutic effects of these drugs. Psychedelic-induced 5HT2A down-regulation is presumably temporary - and if all we need to do is to knock out 5HT2A, it would surely be easiest to just use an antagonist...
Vollenweider FX, & Kometer M (2010). The neurobiology of psychedelic drugs: implications for the treatment of mood disorders. Nature Reviews Neuroscience, 11 (9), 642-51 PMID: 20717121 Posted in
antidepressants,
drugs,
freud,
history,
mental health,
papers




