

Here at Neuroskeptic we have closely followed the development of fMRI scanning on fish.
But a new study has taken it to the next level by scanning... some cheese.
OK, this is not quite true. The study used NMR spectroscopy to analyze the chemistry of some cheeses, in order to measure the effects of different kinds of probiotic bacteria on the composition of the cheese. NMR is the same technology as MRI, and indeed you can use an MRI scanner to gather NMR spectra.
In fact, NMR is Nuclear Magnetic Resonance and MRI is Magnetic Resonance Imaging; it was originally called NMRI, but they dropped the "N" because people didn't like the idea of being scanned by a "nuclear" machine. However, this study didn't actually involve putting cheese into an MRI scanner.
But the important point is that they could have done it by doing that. And if you did that, what with the salmon and now the cheese, you could get a nice MRI-based meal going. All we need is for someone to scan some vegetables, some herbs, and a slice of lemon, and we'd have a delicious dataset. Mmm.
How to cook it? Well, it's actually possible to heat stuff up with an MRI scanner. When scanning people, you set it up to make sure this doesn't happen, but the average fMRI experiment still causes mild heating. It's unavoidable.
I'm not sure what the maximum possible heating effect of an average MRI scanner would be. I doubt anyone has gone out of their way to try and maximize it, but maybe someone ought to look into it. Think of the possibilites.
You've just finished a hard day's scanning and you're really hungry, but the microwave at the MRI building is broken. Not to worry! Just pop your fillet of salmon in probiotic cheese sauce in the magnet, and scan it 'till it's done. You could inspect the images and the chemical composition of the meal before you eat it, to make sure it's just right.
Just make sure you don't use a steel saucepan...
First Fish, Now Cheese, Get Scanned
 01.15
01.15
 wsn
wsn
The Mystery of Stiff Person Syndrome
11.30
wsn
"Stiff Person Syndrome" (SPS) is a rare neurological disease with a silly name but serious symptoms. Not in fact a disorder caused by an overdose of Viagra, the defining feature of SPS is uncontrollable muscle rigidity, which comes and goes in bouts, but generally gets worse over time. However, other symptoms are seen including depression, anxiety, and other neurological features such as cerebellar ataxia.
Not in fact a disorder caused by an overdose of Viagra, the defining feature of SPS is uncontrollable muscle rigidity, which comes and goes in bouts, but generally gets worse over time. However, other symptoms are seen including depression, anxiety, and other neurological features such as cerebellar ataxia.
What causes SPS? Well, it's been known for over 20 years that most SPS patients have antibodies against the enzyme GAD65, which is required for the production of GABA, the main inhibitory neurotransmitter in the brain. The body shouldn't be producing antibodies against its own proteins, but unfortunately this does happen quite often, for various reasons, and the result is autoimmune diseases.
So this all seems to make sense. We know that GABA causes muscle relaxation by reducing the brain's input to the muscles. This is why GABA drugs like Valium are muscle-relaxants, and it's part of the reason why drunk people tend to stagger around.
This also explains the anxiety symptoms, because Valium and beer make you less anxious, while drugs that block GABA cause panic attacks. Anti-GAD65 antibodies block GAD, so less GABA gets made. So SPS is autoimmunity against GAD65. Mystery solved?
Not quite. Anti-GAD65 antibodies are also seen in most people with Type I diabetes, but the vast majority of diabetics luckily don't suffer SPS. Mystery remains.
Two studies just out investigated exactly what the antibodies produced by SPS patients do. Geis et al purified the antibodies from a 53 year old woman with SPS and serious anxiety, and injected them into the brains of some rats.
The rats became very anxious. Here's what the cowardly critters did in a standard rodent anxiety test: they avoided the open spaces, which are naturally scary to rodents, who prefer dark, enclosed places: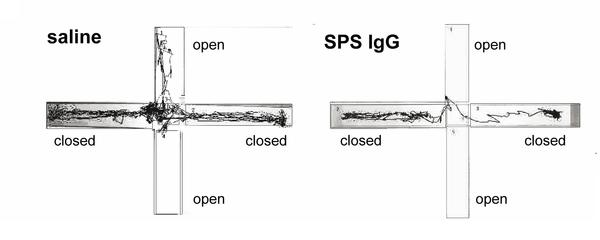 This was associated with reduced GABA production.
This was associated with reduced GABA production.
Meanwhile Manto et al found that anti-GAD65 antibodies from another patient with SPS caused very different effects in rat brains compared to the antibodies derived from a patient with autoimmune cerebellar ataxia, but no SPS symptoms. They also found that two kinds of off-the-shelf anti-GAD65 antibodies commonly used in research had different effects as well.
Taken together this all suggests that SPS is caused by anti-GAD65 antibodies, but they have to be a particular type. Different antibodies cause different symptoms even though they all bind to GAD65.
Presumably this is because it's a big protein, and antibodies could bind to any part of it. Only ones that block the "business end" - the part which actually catalyzes the formation of GABA - will cause problems. A bit like how if you get shot in the heart, that's the end of you, but get shot in the foot and it probably won't be.
Manto MU, Hampe CS, Rogemond V, & Honnorat J (2011). Respective implications of glutamate decarboxylase antibodies in stiff person syndrome and cerebellar ataxia. Orphanet journal of rare diseases, 6 (1) PMID: 21294897
 Posted in
animals,
drugs,
mental health,
papers
Posted in
animals,
drugs,
mental health,
papers
Antidepressants Don't Work...In Fish
13.10
wsn
Here at Neuroskeptic fMRI scanning and antidepressants are both big topics.
As I discussed lask week, fish - specifically salmon - are the next big thing in fMRI and the number of salmon brains being scanned is growing at a remarkable rate. But fish haven't made much of an entrance into the world of antidepressants...until now.
Swedish scientists Holmberg et al have just published a paper asking: Does waterborne citalopram affect the aggressive and sexual behaviour of rainbow trout and guppy?
SSRI antidepressants, of which citalopram is one, are very popular. So popular, in fact, that non-trivial levels of SSRIs have been found in sewage and there's a concern that they might make their way into lakes and rivers and thereby affect the behaviour of the animals living there.
Holmberg et al set out to see what citalopram did to some fish in an attempt to find out whether this is likely to be a major problem. So they put some citalopram in the fish's water supplies and then tested their aggressiveness and also their sex drives. It turns out that one of the main ways of measure fish aggression is to put a mirror in their tank and see if they try to fight their own reflection. Fish are not very bright, really.
Anyway, the good news for fish everywhere was that seven days of citalopram exposure had no effect at all, even at doses much higher than those reported as a pollutant (the maximum dose was 0.1 mg/l). And the authors had no conflicts of interest: Big Pharma had nothing to do with this research, although Big Fish Farmer did because they bought the fish from one.
However, this may not be the end of the story, because it turned out that citalopram was very poorly absorbed into the fish's bloodstreams. But other antidepressants have been reported to accumulate in fish. Clearly, the only way to find out for sure what's going on would be to use fMRI...
Posted in
animals,
antidepressants,
drugs,
fMRI,
funny,
mental health,
papers
fMRI Scanning Salmon - Seriously.
08.12
wsn
Back in 2009, a crack team of neuroscientists led by Craig Bennett (blog) famously put a dead fish into an MRI scanner and showed it some pictures.
They found some blobs of activation - when they used an inappropriately lenient statistical method. Their point, of course, was to draw attention to the fact that you really shouldn't use that method for fMRI. You can read the whole paper here. The Atlantic Salmon who heroically volunteered for the study was no more than a prop. In fact, I believe he ended up getting eaten.
But now, a Japanese team have just published a serious paper which actually used fMRI to measure brain activity in some salmon: Olfactory Responses to Natal Stream Water in Sockeye Salmon by BOLD fMRI.
How do you scan a fish? Well, like this: A total of 6 fish were scanned. The salmon were immobilized by adding an anaesthetic (eugenol) and a muscle relaxant (gallamine) to their tank of water. Then, they were carefully clamped into place to make sure they really wouldn't move, while a stream of oxygenated water was pumped through their tank.
A total of 6 fish were scanned. The salmon were immobilized by adding an anaesthetic (eugenol) and a muscle relaxant (gallamine) to their tank of water. Then, they were carefully clamped into place to make sure they really wouldn't move, while a stream of oxygenated water was pumped through their tank.
Apart from that, it was pretty much a routine fMRI scan.
Why would you want to scan a fish? This is where the serious science comes in. Salmon are born in rivers but they swim out to live in the ocean once they reach maturity. However, they return to the river to breed. What's amazing is that salmon will return to the same river that they were born in - even if they have to travel thousands of miles to get there.
How they manage this is unclear, but the smell (or maybe taste) of the water from their birth river has long been known to be crucial at least once they've reached the right general area (see here for a good overview). Every river contains a unique mixture of chemicals, both natural and artificial (pollutants). Salmon seem to be attracted to whatever chemicals were present in the water when they were young.
In this study, the fMRI revealed that relative to pure water, home-stream water activated a part of the salmon's telencephalon - the most "advanced" part (in humans, it constitutes the vast majority of the brain; in fish, it's tiny). By contrast, a control scent (the amino acid L-serine) did not activate this area, even though the concentration of L-serine was far higher than that of anything in the home-stream water. How this happens is unclear, but further studies of the identified telencephalon area ought to shed more light on it.
So fishMRI is clearly a fast-developing area of neuroscience. In fact, as this graph shows, it's enjoying exponential growth and, if current trends continue, could become almost as popular as scanning people... Link: Also blogged at NeuroDojo.
Link: Also blogged at NeuroDojo.
Bandoh H, Kida I, & Ueda H (2011). Olfactory Responses to Natal Stream Water in Sockeye Salmon by BOLD fMRI. PloS one, 6 (1) PMID: 21264223XMRV - Innocent on All Counts?
11.50
wsn
A bombshell has just gone off in the continuing debate over XMRV, the virus that may or may not cause chronic fatigue syndrome. Actually, 4 bombshells. A set of papers out today in Retrovirology (1,2,3,4) claim that many previous studies claiming to have found the virus haven't actually been detecting XMRV at all.
A set of papers out today in Retrovirology (1,2,3,4) claim that many previous studies claiming to have found the virus haven't actually been detecting XMRV at all.
Here's the rub. XMRV is a retrovirus, a class of bugs that includes HIV. Retroviruses are composed of RNA, but they can insert themselves into the genetic material of host cells as DNA. This is how they reproduce: once their DNA is part of the host cell's chromosomes, that cell is ends up making more copies of the virus.
But there are lots of retroviruses out there, and there used to be yet others that are now extinct. So bits of retroviral DNA are scattered throughout the genome of animals. These are called endogenonous retro-viruses (ERVs).
XMRV is extremely similar to certain ERVs found in the DNA of mice. And mice are the most popular laboratory mammals in the world. So you can see the potential problem: laboratories all over the world are full of mice, but mouse DNA might show up as "XMRV" DNA on PCR tests.
Wary virologists take precautions against this by checking specifically for mouse DNA. But most mouse-contamination tests are targeted at mouse mitochondrial DNA (mtDNA). In theory, a test for mouse mtDNA is all you need, because mtDNA is found in all mouse cells. In theory.
Now the four papers (or are they the Four Horsemen?) argue, in a nutshell, that mouse DNA shows up as "XMRV" on most of the popular tests that have been used in the past, that mouse contamination is very common - even some of the test kits are affected! - and that tests for mouse mtDNA are not good enough to detect the problem.
- Hue et al say that "Taqman PCR primers previously described as XMRV-specific can amplify common murine ERV sequences from mouse suggesting that mouse DNA can contaminate patient samples and confound specific XMRV detection." They go on to show that some human samples previously reported as infected with XMRV, are actually infected with a hybrid of XMRV and a mouse ERV which we know can't infect humans.
- Sato et al report that PCR testing kits from Invitrogen, a leading biotech company, are contaminated with mouse genes including an ERV almost identical to XMRV, and that this shows up as a false positive using commonly used PCR primers "specific to XMRV".
- Oakes et al say that in 112 CFS patients and 36 healthy control, they detected "XMRV" in some samples but all of these samples were likely contaminated with mouse DNA because "all samples that tested positive for XMRV and/or MLV DNA were also positive for the highly abundant IAP long terminal repeat [found only in mice] and most were positive for murine mitochondrial cytochrome oxidase sequences [found only in mice]"
- Robinson et al agree with Oakes et al: they found "XMRV" in some human samples, in this case prostate cancer cells, but they then found that all of the "infected" samples were contaminated with mouse DNA. They recommend that in future, samples should be tested for mouse genes such as the IAP long terminal repeat or cytochrome oxidase, and that researchers should not rely on tests for mouse mtDNA.
I lack the technical knowledge to evaluate these claims, no doubt plenty of people will be rushing to do that before long. (Update: The excellent virologyblog has a more technical discussion of these studies.) But there are a couple of things to bear in mind.
Firstly, these papers cast doubt on tests using PCR to detect XMRV DNA. However, they don't have anything to say about studies which have looked for antibodies against XMRV in human blood, at least not directly. There haven't been many of these, but the paper which started the whole story, Lombardi et al (2009), did look for, and found, anti-XMRV immunity, and also used various other methods to support the idea that XMRV is present in humans. So this isn't an "instant knock-out" of the XMRV theory, although it's certainly a serious blow.
Secondly, if the 'mouse theory' is true, it has serious implications for the idea that XMRV causes chronic fatigue syndrome and also for the older idea that it's linked to prostate cancer. But it still leaves a mystery: why were the samples from CFS or prostate cancer patients more likely to be contaminated with mouse DNA than the samples from healthy controls?
Robert A Smith (2010). Contamination of clinical specimens with MLV-encoding nucleic acids: implications for XMRV and other candidate human retroviruses Retrovirology : 10.1186/1742-4690-7-112Cannabinoids in Huntington's Disease
05.10
wsn
Two recent papers have provided strong evidence that the brain's endocannabinoid system is dysfunctional in Huntington's Disease, paving the way to possible new treatments. Huntington's Disease is a genetic neurological disorder. Symptoms generally appear around age 40, and progress gradually from subtle movement abnormalities to dementia and complete loss of motor control. It's incurable, although medication can mask some of the symptoms. Singer Woodie Guthrie is perhaps the disease's best known victim: he ended his days in a mental institution.
Huntington's Disease is a genetic neurological disorder. Symptoms generally appear around age 40, and progress gradually from subtle movement abnormalities to dementia and complete loss of motor control. It's incurable, although medication can mask some of the symptoms. Singer Woodie Guthrie is perhaps the disease's best known victim: he ended his days in a mental institution.
The biology of Huntington's is only partially understood. It's caused by mutations in the huntingtin gene, which lead to the build-up of damaging proteins in brain cells, especially in the striatum. But exactly how this produces symptoms is unclear.
The two new papers show that cannabinoids play an important role. First off, Van Laere et al used PET imaging to measure levels of CB1 receptors in the brain of patients in various stages of Huntington's. CB1 is the main cannabinoid receptor in the brain; it responds to natural endocannabinoid neurotransmitters, and also to THC, the active ingredient in marijuana.
They found serious reductions in all areas of the brain compared to healthy people, and interestingly, the loss of CB1 receptors occurred early in the course of the disease: That was an important finding, but it didn't prove that CB1 loss was causing any problems: it might have just been a side-effect of the disease. Now another study using animals has shown that it's not: Blazquez et al. They studied mice with the same mutation that causes Huntington's in humans. These unfortunate rodents develop Huntington's, unsurprisingly.
That was an important finding, but it didn't prove that CB1 loss was causing any problems: it might have just been a side-effect of the disease. Now another study using animals has shown that it's not: Blazquez et al. They studied mice with the same mutation that causes Huntington's in humans. These unfortunate rodents develop Huntington's, unsurprisingly.
They found that Huntington's mice who also had a mutation eliminating the CB1 receptor suffered more severe symptoms, which appeared earlier, and progressed faster. This suggests that CB1 plays a neuroprotective role, which is consistent with lots of earlier studies in other disorders.
If so, drugs that activate CB1 - like THC - might be able to slow down the progression of the disease, and indeed it did: Huntington's mice given THC injections stayed healthier for longer, although they eventually succumbed to the disease. Further experiments showed that mutant huntingtin switches off expression of the CB1 receptor gene, explaining the loss of CB1.
This graph shows performance on the RotaRod test of co-ordination: mice with Huntington's (R6/2) got worse and worse starting at 6 weeks of age (white bars), but THC slowed down the decline (black bars). The story was similar for other symptoms, and for the neural damage seen in the disease. They conclude that:
They conclude that:
Altogether, these results support the notion that downregulation of type 1 cannabinoid receptors is a key pathogenic event in Huntington’s disease, and suggest that activation of these receptors in patients with Huntington’s disease may attenuate disease progression.Now, this doesn't mean people with Huntington's should be heading out to buy Bob Marley posters and bongs just yet. For one thing, Huntington's disease often causes psychiatric symptoms, including depression and psychosis. Cannabis use has been linked to psychosis fairly convincingly, so marijuana might make those symptoms worse.
Still, it's very promising. In particular, it will be interesting to try out next-generation endocannabinoid boosting drugs, such as FAAH inhibitors, which block the breakdown of anandamide, one of the most important endocannabinoids.
In animals FAAH inhibitors have pain relieving, anti-anxiety, and other beneficial effects, but they don't cause the same behavioural disruptions that THC does. This suggests that they wouldn't get people high, either, but there's no published data on what they do in humans yet...
Van Laere K, et al. (2010). Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. Journal of nuclear medicine : official publication, Society of Nuclear Medicine, 51 (9), 1413-7 PMID: 20720046Blázquez C, et al. (2010). Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain : a journal of neurology PMID: 20929960
Posted in
animals,
CNR1,
drugs,
mental health,
papers
This Is Your Brain's Anti-Drug
08.10
wsn
 What's your anti-drug? Well, it might well be hemopressin. At least, that's probably your anti-marijuana.
What's your anti-drug? Well, it might well be hemopressin. At least, that's probably your anti-marijuana.
Hemopressin is a small protein that was discovered in the brains of rodents in 2003: its name comes from the fact that it's a breakdown product of hemoglobin and that it can lower blood pressure.
No-one seems to have looked to see whether hemopressin is found in humans, yet, but it seems very likely. Almost everything that's in your brain is in a mouse's brain, and vice versa.
Pharmacologically, hemopressin's literally an anti-marijuana molecule: it's an inverse agonist at CB1 receptors, which are the ones targeted by the psychoactive compounds in marijuana, and also by the neurotransmitters known as endocannabinoids. Cannabinoids turn CB1 receptors on, hemopressin turns them off.
Artificial CB1 blockers were developed as weight loss drugs, and one of them, rimonabant, made it onto the market - but it was banned after it turned out that it caused depression and anxiety in many people.
So hemopressin is Nature's rimonabant: in which case, it ought to do what rimonabant does, which is to reduce appetite. And indeed a Journal of Neuroscience paper just out from Godd et al shows that it does just that, in rats and mice: injections of hemopressin reduced feeding.
Interestingly, this worked even when it was injected by the standard route under the skin - many proteins can't enter the brain if they're given this way, because they can't cross the blood-brain barrier, meaning that they have to be injected directly into the brain, which makes researching them much harder. So hemopressin, with any luck, will be pretty easy to study. Any volunteers for the first human trial...?
Do It Like You Dopamine It
14.40
wsn
Neuroskeptic readers will know that I'm a big fan of theories. Rather than just poking around (or scanning) the brain under different conditions and seeing what happens, it's always better to have a testable hypothesis. I just found a 2007 paper by Israeli computational neuroscientists Niv et al that puts forward a very interesting theory about dopamine. Dopamine is a neurotransmitter, and dopamine cells are known to fire in phasic bursts - short volleys of spikes over millisecond timescales - in response to something which is either pleasurable in itself, or something that you've learned is associated with pleasure. Dopamine is therefore thought to be involved in learning what to do in order to get pleasurable rewards.
I just found a 2007 paper by Israeli computational neuroscientists Niv et al that puts forward a very interesting theory about dopamine. Dopamine is a neurotransmitter, and dopamine cells are known to fire in phasic bursts - short volleys of spikes over millisecond timescales - in response to something which is either pleasurable in itself, or something that you've learned is associated with pleasure. Dopamine is therefore thought to be involved in learning what to do in order to get pleasurable rewards.
But baseline, tonic dopamine levels vary over longer periods as well. The function of this tonic dopamine firing, and its relationship, if any, to phasic dopamine signalling, is less clear. Niv et al's idea is that the tonic dopamine level represents the brain's estimate of the average availability of rewards in the environment, and that it therefore controls how "vigorously" we should do stuff.
A high reward availability means that, in general, there's lots of stuff going on, lots of potential gains to be made. So if you're not out there getting some reward, you're missing out. In economic terms, the opportunity cost of not acting, or acting slowly, is high - so you need to hurry up. On the other hand, if there's only minor rewards available, you might as well take things nice and slow, to conserve your energy. Niv et al present a simple mathematical model in which a hypothetical rat must decide how often to press a lever in order to get food, and show that it accounts for the data from animal learning experiments. The distinction between phasic dopamine (a specific reward) vs. tonic dopamine (overall reward availability) is a bit like the distinction between fear vs. anxiety. Fear is what you feel when something scary, i.e. harmful, is right there in front of you. Anxiety is the sense that something harmful could be round the next corner.
The distinction between phasic dopamine (a specific reward) vs. tonic dopamine (overall reward availability) is a bit like the distinction between fear vs. anxiety. Fear is what you feel when something scary, i.e. harmful, is right there in front of you. Anxiety is the sense that something harmful could be round the next corner.
This theory accounts for the fact that if you give someone a drug that increases dopamine levels, such as amphetamine, they become hyperactive - they do more stuff, faster, or at least try to. That's why they call it speed. This happens to animals too. Yet this hyperactivity starts almost immediately, which means that it can't be a product of learning.
It also rings true in human terms. The feeling that everything's incredibly important, and that everyday tasks are really exciting, is one of the main effects of amphetamine. Every speed addict will have a story about the time they stayed up all night cleaning every inch of their house or organizing their wardrobe. This can easily develop into the compulsive, pointless repetition of the same task over and over. People with bipolar disorder often report the same kind of thing during (hypo)mania.
What controls tonic dopamine levels? A really brilliantly elegant answer would be: phasic dopamine. Maybe every time phasic dopamine levels spike in response to a reward (or something which you've learned to associate with a reward), some of the dopamine gets left over. If there's lots of phasic dopamine firing, which suggests that the availability of rewards is high, the tonic dopamine levels rise.
Unfortunately, it's probably not that simple, as signals from different parts of the brain seem to alter tonic and phasic dopamine firing largely independently, and this would mean that tonic dopamine would only increase after a good few rewards, not pre-emptively, which seems unlikely. The truth is, we don't know what sets the dopamine tone, and we don't really know what it does; but Niv et al's account is the most convincing I've come across...
Posted in
animals,
drugs,
mental health,
papers
Mice That Fight for Their Rights
15.10
wsn
Israeli biologists Feder et al report on Selective breeding for dominant and submissive behavior in Sabra mice. Mice are social animals and like many species, they show dominance hierarchies. When they first meet, they'll often fight each other. The winner gets to be Mr (or Mrs) Big, and they enjoy first pick of the food, mating opportunities, etc - for as long as they can remain dominant.
Mice are social animals and like many species, they show dominance hierarchies. When they first meet, they'll often fight each other. The winner gets to be Mr (or Mrs) Big, and they enjoy first pick of the food, mating opportunities, etc - for as long as they can remain dominant.
But what determines which mice become top dog... ? Feder et al show that it's partially under genetic control. They took a normal population of laboratory mice, paired them up, and made them battle for supremacy in a simple set-up in which only one mouse can get access to a central food supply: At first, only about 30% of pairs developed clear dominance/submission relationships, but the ones that did were selectively bred: dominant males mated with dominant females, and submissive males with submissive females. The offspring were put through the same process, and it was repeated.
At first, only about 30% of pairs developed clear dominance/submission relationships, but the ones that did were selectively bred: dominant males mated with dominant females, and submissive males with submissive females. The offspring were put through the same process, and it was repeated.
The results were dramatic: After 4 generations of successive selection, 80% of the pairs showed clear dominance and submission behaviour. And with each generation of breeding, the dominance relationships appeared faster, and stronger: at first the winners only got slightly more access to the food, but by the 4th generation, they almost completely monopolized it. As expected the mice bred to be dominant were overwhelmingly more likely to end up on top. The differences were not due to general differences in activity levels or anxiety. But the naturally timid mice could be made to fight for their rights by treating them with antidepressants - after a month of imipramine, they were taking crap from no-one.
But the naturally timid mice could be made to fight for their rights by treating them with antidepressants - after a month of imipramine, they were taking crap from no-one.
Feder et al say that previous studies have also shown anti-submissive effects of antidepressants, while drugs used to treat mania reduce dominance. Anyone who's experienced a mood disorder will probably be able to relate to this: depressed people tend to feel like they belong at the bottom of the pecking order of life, while mania is classically associated with believing you're the greatest person in history.
So dominance and submission could provide a useful way of testing the effects of drugs on mood. If so, it would be useful, because current animal models of depression and antidepressants etc. mostly rely on putting animals in a glass of water and seeing how long they take to stop struggling...
Posted in
animals,
antidepressants,
drugs,
funny,
mental health,
papers
Do Cats Hallucinate?
12.40
wsn
I have two cats. One is about four, and he is a psychopath. The other is sixteen - elderly, in cat terms - and I've recently noticed some changes in her behaviour. For one, she's become a lot more affectionate, and she demands constant attention - she meows at people on sight, follows you around, and almost always comes and sits on top of you, or on top of whatever you're doing/reading/typing.
For one, she's become a lot more affectionate, and she demands constant attention - she meows at people on sight, follows you around, and almost always comes and sits on top of you, or on top of whatever you're doing/reading/typing.
But on top of that, she's started pausing in the middle of whatever she's doing and staring at empty corners, or walls. All cats sit down and gaze into space a lot of the time, but this is different - it happens in the middle of normal actions, like eating or walking around. What does this mean?
Could she be hallucinating? Hallucinations are unfortunately not uncommon in elderly people. Seeing and hearing things that aren't there is a major symptom of Alzheimer's, and other forms of dementia. Do cats get Alzheimer's? The internet says: yes. In terms of scientific research there doesn't seem to have been much, but a few studies have found Alzheimer's-like changes (amyloid-beta protein accumulation) in the brains of old cats. Whether these cause the same symptoms as they do in people is unclear, but, why not?
How would you know if an animal was hallucinating? They can't talk about it, and unlike say hunger or pain, they don't have specific ways of communicating it through body language or cries. A hallucinating animal would, presumably, react fairly normally to whatever it thought it saw or heard: so hallucinations would manifest as normal behaviours, but in inappropriate situations. Whether this is what's happening to my cat, I'm not sure, but again, it's possible.
A more philosophical issue is whether we can conclude that this kind of out-of-context behaviour means the animal is experiencing a hallucination. But this is really just the age old question of whether animals have conciousness at all. If they do, then they can presumably hallucinate: if you can be concious of sensations, you can be concious of false sensations.
For what its worth, my view is that animals, at any rate for mammals, are concious. Humans are (although technically we only know for sure that we personally are, and have to assume the same is true of others.) Mammalian brains are structured in a similar way to our own; they're made of the same cells; they use the same neurotransmitters and the same drugs interfere with them in the same ways; pretty much all of the brain regions are there, although the sizes differ.
There's of course a big difference between us and other mammals: we have language, and conceptual thinking, and so forth. But does conciousness depend on that? It seems unlikely, just because most of what we're concious of at any one time isn't anything to do with those specifically human things.
Right now, I'm concious of what I can see, what I can hear, what I can feel with my fingertips, and the thoughts I'm writing down. Only 1/4 of that (to put it crudely) is unique to humans. And I'm not always aware of thoughts or words; there are plenty of times when I'm only aware of sensations and perceptions.
Probably the closest we get to animal conciousness is in strong, primitive experiences like pain, panic and anger, in which we "take leave of our senses" - not meaning that we become unconscious, but that we temporarily stop being able to "think straight" i.e. like a human. That doesn't mean that animals spend all their time in some extreme emotional state, but it's harder for us to know what it's like to be a relaxed cat because generally when we're relaxed, we're thinking (or daydreaming, etc. Although who's to say cats don't? They dream, after all...)
Posted in
animals,
drugs,
philosophy
Absinthe Fact and Fiction
11.53
wsn
 Absinthe is a spirit. It's very strong, and very green. But is it something more?
Absinthe is a spirit. It's very strong, and very green. But is it something more?
I used to think so, until I came across this paper taking a skeptical look at the history and science of the drink, Padosch et al's Absinthism a fictitious 19th century syndrome with present impact
Absinthe is prepared by crushing and dissolving the herb wormwood in unflavoured neutral alcohol and then distilling the result; other herbs and spices are added later for taste and colour.
It became extremely popular in the late 19th century, especially in France, but it developed a reputation as a dangerous and hallucinogenic drug. Overuse was said to cause insanity, "absinthism", much worse than regular alcoholism. Eventually, absinthe was banned in the USA and most but not all European countries.
Much of the concern over absinthe came from animal experiments. Wormwood oil was found to cause hyperactivity and seizures in cats and rodents, whereas normal alcohol just made them drunk. But, Padosch et al explain, the relevance of these experiments to drinkers is unclear, because they involved high doses of pure wormwood extract, whereas absinthe is much more dilute. The fact that authors at the time used the word absinthe to refer to both the drink and the pure extract added to the confusion. It's now known that wormwood, or at least some varieties of it, contains thujone, which can indeed cause seizures, and death, due to being a GABA antagonist. Until a few years ago it was thought that old-style absinthe might have contained up to 260 mg of thujone per litre, a substantial dose.
It's now known that wormwood, or at least some varieties of it, contains thujone, which can indeed cause seizures, and death, due to being a GABA antagonist. Until a few years ago it was thought that old-style absinthe might have contained up to 260 mg of thujone per litre, a substantial dose.
But that was based on the assumption that all of the thujone in the wormwood ended up in the drink prepared from it. Chemical analysis of actual absinthe has repeatedly found that it contains no more than about 6 mg/L thujone. The alcohol in absinthe would kill you long before you drank enough to get any other effects. As the saying goes, "the dose makes the poison", something that is easily forgotten.
As Padosch et al point out, it's possible that there are other undiscovered psychoactive compounds in absinthe, or that long-term exposure to low doses of thujone does cause "absinthism". But there is no evidence for that so far. Rather, they say, absinthism was just chronic alcoholism, and absinthe was no more or less dangerous than any other spirit.
I'm not sure why, but drinks seem to attract more than their fair share of urban myths. Amongst many others I've heard that the flakes of gold in Goldschläger cause cuts which let alcohol into your blood faster; Aftershock crystallizes in your stomach, so if you drink water the morning afterwards, you get drunk again; and that the little worm you get at the bottom of some tequilas apparently contains especially concentrated alcohol, or hallucinogens, or even cocaine maybe.
Slightly more serious is the theory that drinking different kinds of drinks instead of sticking to just one gets you drunk faster, or gives you a worse hangover, or something, especially if you do it in a certain order. Almost everyone I know believes this, although in my drinking experience it's not true, but I'm not sure that it's completely bogus, as I have heard somewhat plausible explanations i.e. drinking spirits alongside beer leads to a concentration of alcohol in your stomach that's optimal for absorption into the bloodstream... maybe.
Link: Not specifically related to this but The Poison Review is an excellent blog I've recently discovered all about poisons, toxins, drugs, and such fun stuff.
Posted in
animals,
bad neuroscience,
drugs,
history,
mental health,
papers
The Sweet Taste of Cannabinoids
07.44
wsn
 Every stoner knows about the munchies, the fondness for junk food that comes with smoking marijuana. Movies have been made about it.
Every stoner knows about the munchies, the fondness for junk food that comes with smoking marijuana. Movies have been made about it.
It's not just that being on drugs makes you like eating: stimulants, like cocaine and amphetamine, decrease appetite. The munchies are something specific to marijuana. But why?
New research from a Japanese team reveals that marijuana directly affects the cells in the taste buds which detect sweet flavours - Endocannabinoids selectively enhance sweet taste.
Yoshida et al studied mice, and recorded the electrical signals from the chorda tympani (CT), which carries taste information from the tongue to the brain.
They found that injecting the mice with two chemicals, 2AG and AEA, markedly increased the strength of the signals produced in response to sweet tastes - such as sugar, or the sweetener saccharine. However, neither had any effect on the strength of the response to other flavours, like salty, bitter, or sour. Mice given endocannabinoids were also more eager to eat and drink sweet things, which confirms previous findings.
2-AG and AEA are both endocannabinoids, an important class of neurotransmitters. Marijuana's main active ingredient, Δ9-THC, works by mimicking the action of endocannabinoids. Although Δ9-THC wasn't tested in this study, it's extremely likely that it has the same effects as 2-AG and AEA. In follow-up experiments, Yoshida et al found that endocannabinoids enhance sweet taste responses by acting on cannabinoid type 1 (CB1) receptors on the tongue's sweet taste cells themselves. In fact, over half of the sweet receptor cells expressed CB1 receptors!
In follow-up experiments, Yoshida et al found that endocannabinoids enhance sweet taste responses by acting on cannabinoid type 1 (CB1) receptors on the tongue's sweet taste cells themselves. In fact, over half of the sweet receptor cells expressed CB1 receptors!
This is an important finding, because CB1 receptors are already known to regulate the pleasurable response to sweet foods (amongst other things) in the brain. These new data don't challenge this, but suggest that CB1 also modulates the most basic aspects of sweet taste perception. The munchies are probably caused by Δ9-THC acting at multiple levels of nervous system.
This paper also sheds light on CB1 antagonists. Given that drugs which activate CB1 make people eat more, it would make sense if CB1 blockers made people eat less, and therefore lose weight, a kind of anti-munchies effect. And indeed they do. Which is why rimonabant, a CB1 antagonist, was released onto the market in 2006 as a weight loss drug. It worked pretty well, although unfortunately it also it caused clinical depression in some people, so it was banned in Europe in 2008 and was never approved in the USA for the same reason.
The depression was almost certainly caused by antagonism at CB1 receptors in the brain, but Yoshida et al's findings suggest that a CB1 antagonist which didn't enter the brain, and only affected peripheral sites such as the taste buds, might be able to make people less fond of sweet foods without causing the same side-effects. Who knows - in a few years you might even be able to buy CB1 antagonist chewing gum to help you stick to your diet...
In the Brain, Acidity Means Anxiety
03.30
wsn
 According to Mormon author and fruit grower "Dr" Robert O. Young, pretty much all diseases are caused by our bodies being too acidic. By adopting an "alkaline lifestyle" to raise your internal pH (lower pH being more acidic), you'll find that
According to Mormon author and fruit grower "Dr" Robert O. Young, pretty much all diseases are caused by our bodies being too acidic. By adopting an "alkaline lifestyle" to raise your internal pH (lower pH being more acidic), you'll find that
if you maintain the saliva and the urine pH, ideally at 7.2 or above, you will never get sick. That’s right you will NEVER get sick!Wow. Important aspects of the alkaline lifestyle include eating plenty of the right sort of fruits and vegetables, ideally ones grown by Young, and taking plenty of nutritional supplements. These don't come cheap, but when the payoff is being free of all diseases, who could complain?
Young calls his amazing theory the Alkavorian Approach™, aka the New Biology™. Almost everyone else calls it quack medicine and pseudoscience. Because it is quack medicine and pseudoscience. But a paper just published in Cell suggests an interesting role for pH in, of all things, anxiety and panic - The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior.
The authors, Ziemann et al, were interested in a protein called Acid Sensing Ion Channel 1a, ASIC1a, which as the name suggests, is acid-sensitive. Nerve cells expressing ASIC1a are activated when the fluid around them becomes more acidic.
One of the most common causes of acidosis (a fall in body pH) is carbon dioxide, CO2. Breathing is how we get rid of the CO2 produced by our bodies; if breathing is impaired, for example during suffocation, CO2 levels rise, and pH falls as CO2 is converted to carbonic acid in the bloodstream.
In previous work, Ziemann et al found that the amygdala contains lots of ASIC1a. This is intriguing, because the amygdala is a brain region believed to be involved in fear, anxiety and panic, although it has other functions as well. It's long been known that breathing air with added CO2 can trigger anxiety and panic, especially in people vulnerable to panic attacks.
What's unclear is why this happens; various biological and psychological theories have been proposed. Ziemann et al set out to test the idea that ASIC1a in the amygdala mediates anxiety caused by CO2.
 In a number of experiments they showed that mice genetically engineered have no ASIC1a (knockouts) were resistant to the anxiety-causing effects of air containing 10% or 20% CO2. Also, unlike normal mice, the knockouts were happy to enter a box with high CO2 levels - normal mice hated it. Injections of a weakly acidic liquid directly into the amygdala caused anxiety in normal mice, but not in the knockouts.
In a number of experiments they showed that mice genetically engineered have no ASIC1a (knockouts) were resistant to the anxiety-causing effects of air containing 10% or 20% CO2. Also, unlike normal mice, the knockouts were happy to enter a box with high CO2 levels - normal mice hated it. Injections of a weakly acidic liquid directly into the amygdala caused anxiety in normal mice, but not in the knockouts. Most interestingly, they found that knockout mice could be made to fear CO2 by giving them ASIC1a in the amygdala. Knockouts injected in the amygdala with a virus containing ASIC1a DNA, which caused their cells to start producing the protein, showed anxiety (freezing behaviour) when breathing CO2. But it only worked if the virus was injected into the amygdala, not nearby regions.
Most interestingly, they found that knockout mice could be made to fear CO2 by giving them ASIC1a in the amygdala. Knockouts injected in the amygdala with a virus containing ASIC1a DNA, which caused their cells to start producing the protein, showed anxiety (freezing behaviour) when breathing CO2. But it only worked if the virus was injected into the amygdala, not nearby regions.This is a nice series of experiments which shows convincingly that ASIC1a mediates acidosis-related anxiety, at least in mice. What's most interesting however is that it also seems to involved in other kinds of anxiety and fear. The ASIC1a knockout mice were slightly less anxious in general; injections of an alkaline solution prevented CO2-related anxiety, but also reduced anxiety caused by other scary things, such as the smell of a cat.
The authors conclude by proposing that amygdala pH might be involved in fear more generally
Thus, we speculate that when fear-evoking stimuli activate the amygdala, its pH may fall. For example, synaptic vesicles release protons, and intense neural activity is known to lower pH.But this is, as they say, speculation. The link between CO2, pH and panic attacks seems more solid. As the authors of another recent paper put it
We propose that the shared characteristics of CO2/H+ sensing neurons overlap to a point where threatening disturbances in brain pH homeostasis, such as those produced by CO2 inhalations, elicit a primal emotion that can range from breathlessness to panic.
Ziemann, A., Allen, J., Dahdaleh, N., Drebot, I., Coryell, M., Wunsch, A., Lynch, C., Faraci, F., Howard III, M., & Welsh, M. (2009). The Amygdala Is a Chemosensor that Detects Carbon Dioxide and Acidosis to Elicit Fear Behavior Cell, 139 (5), 1012-1021 DOI: 10.1016/j.cell.2009.10.029 Posted in
amygdala,
animals,
mental health,
papers,
woo
Deep Brain Stimulation for Depressed Rats
13.40
wsn
Deep-brain stimulation (DBS) is probably the most exciting emerging treatment in psychiatry. DBS is the use of high-frequency electrical current to alter the function of specific areas of the brain. Originally developed for Parkinson's disease, over the past five years DBS has been used experimentally in severe clinical depression, OCD, Tourette's syndrome, alcoholism, and more. Reports of the effects have frequently been remarkable, but there have been few scientifically rigorous studies, and the number of psychiatric patients treated to date is just dozens. So the true usefulness of the technique is unclear. How DBS works is also a mystery. Even the most basic questions - such as whether high-frequency stimulation switches the brain "on" or "off" - are still being debated.
Reports of the effects have frequently been remarkable, but there have been few scientifically rigorous studies, and the number of psychiatric patients treated to date is just dozens. So the true usefulness of the technique is unclear. How DBS works is also a mystery. Even the most basic questions - such as whether high-frequency stimulation switches the brain "on" or "off" - are still being debated.
Recent data from rodents sheds some important light on the issue: Antidepressant-Like Effects of Medial Prefrontal Cortex Deep Brain Stimulation in Rats. The authors took rats, and implanted DBS electrodes in the infralimbic cortex. This area is part of the vmPFC. It's believed to be the rat equivalent of the human region BA25, the subgenual cingulate cortex, which is the most common target for DBS in depression. The current settings (100 microA, 130 Hz, 90 microsec) were chosen to be similar to the ones used in humans. In a standard rat model of depression, the forced-swim test, infralimbic DBS exerted antidepressant-like effects. DBS was equally as effective as imipramine, a potent antidepressant, in terms of reducing "depression-like" behaviours, namely immobility.
In a standard rat model of depression, the forced-swim test, infralimbic DBS exerted antidepressant-like effects. DBS was equally as effective as imipramine, a potent antidepressant, in terms of reducing "depression-like" behaviours, namely immobility.
This is not all that surprising. Almost everything which treats depression in humans also reduces immobility in this test (along with few things which don't treat it). Much more interesting is what did and did not block the effects of DBS in these rats.
First off, DBS worked even when the rat's infralimbic cortex had been destroyed by the toxin ibotenic acid. This strongly suggests that DBS does not work simply by activating the infralimbic cortex, even though this is where the electrodes were implanted.
Crucially, infralimbic lesions did not have an antidepressant effect per se, which also rules out the theory that DBS works by inactivating this region. (Infralimbic lesions produced by other methods did have a mild antidepressant effect, but it was smaller than the effect of DBS. This may still be important, however.) What did block the effects of DBS was the depletion of serotonin (5HT). Serotonin is known to its friends as the brain's "happy chemical", although it's a bit more complicated than that. Most antidepressants target serotonin. And rats whose serotonin systems had been lesioned got no benefit from DBS in this study.
What did block the effects of DBS was the depletion of serotonin (5HT). Serotonin is known to its friends as the brain's "happy chemical", although it's a bit more complicated than that. Most antidepressants target serotonin. And rats whose serotonin systems had been lesioned got no benefit from DBS in this study.
So this suggests that DBS might work by affecting serotonin, and indeed, DBS turned out to greatly increase serotonin release, even in a distant part of the brain (the hippocampus). Interestingly this lasted for nearly two hours after the electrodes were switched off. Depletion of another neurotransmitter, noradrenaline, did not alter the effects of DBS.
Depletion of another neurotransmitter, noradrenaline, did not alter the effects of DBS.
Overall, it seems that infralimbic DBS works by increasing serotonin release, but that this is not because it activates or inactivates the infralimbic cortex itself. Rather, nearby structures must be involved. The most likely explanation is that DBS affects nearby white-matter tracts carrying signals between other areas of the brain; the infralimbic cortex might just happen to be "by the roadside". Many researchers believe that this is how DBS works in humans, but this is the first hard evidence for this.
Of course, evidence from rats is never all that hard when it comes to human mental illness. We need to know whether the same thing is true in people. As luck would have it, you can temporarily reduce human serotonin levels with a technique called acute tryptophan depletion This reverses the effects of antidepressants in many people. If this rat data is right, it should also temporarily reverse the benefits of DBS. Someone should do this experiment as soon as possible - I'd like to do it myself, but I'm British, and all the DBS research happens in America. Bah, humbug, old bean.
There's a couple of others things to note here. In other behavioural tests, infralimbic DBS also had antidepressant-like effects: it seemed to reduce anxiety, and it made rats more resistant to the stress of having electrical shocks (although only slightly.) Finally, DBS in another region, the striatum, had no antidepressant effect at all. That's a bit odd because DBS of the striatum does seem to treat depression in humans - but the part of the striatum targeted here, the caudate-putamen, is quite separate to the one targeted in human depression, the nucleus accumbens.
Posted in
animals,
antidepressants,
dbs,
mental health,
papers,
vmPFC





{kind=link}