

 12.25
12.25
 wsn
wsn
A team of Japanese scientists have found the most sarcastic part of the brain known to date. They also found the metaphor centre of the brain and, well, it's kind of like a pair of glasses. The paper is Distinction between the literal and intended meanings of sentences and it's brought to you by Uchiyama et al. They took 20 people and used fMRI to record neural activity while the volunteers read 4 kinds of statements:
The paper is Distinction between the literal and intended meanings of sentences and it's brought to you by Uchiyama et al. They took 20 people and used fMRI to record neural activity while the volunteers read 4 kinds of statements:
- Literally true
- Nonsensical
- Sarcastic
- Metaphorical
Here's what they found. Compared to the literally-true and the nonsensical statements, which were a control condition, metaphorical statements activated the head of the caudate nucleus, the thalamus, and an area of the medial PFC they dub the "arMPFC" but which other people might call the pgACC or something even more exotic; names get a bit vague in the frontal lobe.

The caudate nucleus, as I said, looks like a pair of glasses. Except without the nose bit. The area activated by metaphors was the "lenses". Kind of.
Sarcasm however activated the same mPFC region, but not the caudate:
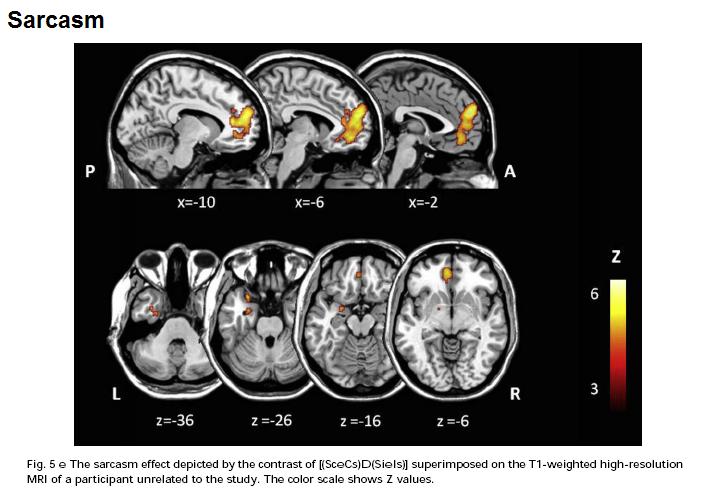 Sarcasm also activated the amygdala.
Sarcasm also activated the amygdala.*
So what? This is a very nice fMRI study. 20 people is a lot, the task was well-designed and the overlap of the mPFC blobs in the sarcasm-vs-control and the metaphor-vs-control tasks was impressive. There's clearly something going on there in both cases, relative to just reading literal statements. Something's going on in the caudate and thalamus with metaphor but not sarcasm, too.
But what can this kind of study tell us about the brain? They've localized something-about-metaphor to the caudate nucleus, but what is it, and what does the caudate actually do to make that thing happen?
The authors offer a suggestion - the caudate is involved in "searching for the meaning" of the metaphorical statement in order to link it to the context, and work out what the metaphor is getting at. This isn't required for sarcasm because there's only one, literal, meaning - it's just reversed, the speaker actually thinks the exact opposite. Whereas with both sarcasm and metaphor you need to attribute intentions (mentalizing or "Theory of Mind").
But what can this kind of study tell us about the brain? They've localized something-about-metaphor to the caudate nucleus, but what is it, and what does the caudate actually do to make that thing happen?
The authors offer a suggestion - the caudate is involved in "searching for the meaning" of the metaphorical statement in order to link it to the context, and work out what the metaphor is getting at. This isn't required for sarcasm because there's only one, literal, meaning - it's just reversed, the speaker actually thinks the exact opposite. Whereas with both sarcasm and metaphor you need to attribute intentions (mentalizing or "Theory of Mind").
That's as plausible an account as any but the problem is that we have no way of knowing, at least not from imaging studies, if it's true or not. As I said this is not the fault of this study but rather an inherent challenge for the whole enterprise. The problem is - switch on your caudate, metaphor coming up - a lot like the challenge facing biology in the aftermath of the Human Genome Project.
The HGP mapped the human genome, and like any map it told us where stuff is, in this case where genes are on chromosomes. You can browse it here. But by itself this didn't tell us anything about biology. We still have to work out what most of these genes actually do; and then we have to work out how they interact; and they we have to work out how those interactions interact with other genes and the environment...
Genomics people call this, broadly speaking, "annotating" the genome, although this is not perhaps an ideal term because it's not merely scribbling notes in the margins, it's the key to understanding. Without annotation, the genome's just a big list.
fMRI is building up a kind of human localization map, a blobome if you will, but by itself this doesn't really tell us much; other tools are required.
 Posted in
Posted in 























{kind=link}